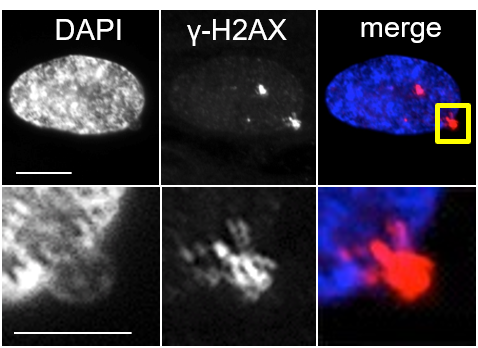
We first characterized Cytoplasmic Chromatin Fragments (CCF) in senescent cells and in collaboration defined a role for CCF as drivers of inflammation via the cGAS/STING cytoplasmic DNA sensing anti-viral pathway. Cellular senesence is a potent tumor suppressor mechanism by virtue of proliferation arrest and the senescence associated secretory phenotype (SASP) which promotes clearance of pre-malignant cells by the immune system. However, the mechanism responsible for initiation of SASP has been unknown. In 2013, my lab first characterized and named CCF as fragments of chromatin expelled from the nucleus of senescent cells into the cytoplasm (Ivanov et al., 2013). Then, in 2015, in collaboration with Shelley Berger's laboratory, we showed that formation of CCF depends on interaction of lamin B1 and autophagy adaptor LC3 in the nucleus, and that lamin B1 is a nuclear substrate of autophagy (Dou et al., 2015). Most recently, again with Shelley Berger's lab, we have shown that CCF are sensed by the cytoplasmic DNA sensing anti-viral apparatus, cGAS and STING, and this leads to activation of NFkB and SASP in senescent cells (Dou et al., 2017). The role of CCF as triggers of SASP, via cGAS and STING, has been confirmed by several other labs.
- Ivanov, A., Pawlikowski, J., Manoharan, I., van Tuyn, J., Nelson, D. M., Rai, T. S., Shah, P. P., Drotar, M., Wu, H., Berger, S. L., and Adams, P.D. (2013) Lysosome-dependent processing of histones in senescent cells. J. Cell Biol., 202:129-43. doi: 10.1083/jcb.201212110.
- Dou, Z., Capell, BC., Drake, AM., Shah, PP., Dorsey, J., Simola, D., Donahue, G., Zhu, Z., Sammons, M., Rai, TS., Natale, C., Ridky, T., Goldman, R., Adams, P.D., Berger, SL. (2015) Autophagy mediates degradation of nuclear lamina. Nature, 527:105-9. doi: 10.1038/nature15548. Epub 2015 Oct 28.
- Dou, D., Ghosh, K., Vizioli, MG., Zhu, J., Sen, P., Wangensteen, KJ., Simithy, J., Lan, Y., Lin, Y., Zhou, Z., Capell, BC., Xu, C., Xu, M., Kieckhaefer, JE., Jiang, T., Shoshkes-Carmel, M., Ahasan Al Tanim, KM., Barber, GN., Seykora, JT., Millar, SE., Kaestner, KH., Garcia, BA., Adams, P.D.*, Berger, SL*. (2017) Cytoplasmic chromatin triggers inflammation in senescence and cancer. *Corresponding authors. Nature, 550 : 402-406. doi: 10.1038/nature24050.
- Vizioli, M.G., Liu, T., Miller, K.N., Robertson, N., Gilroy, K., Lagnado, A., Perez-Garcia, A., Kiourtis, C., Dasgupta, N., Lei, X., Kruger, P.J., Nixon, C., Clark, W., Jurk, D., Bird, T.G., Passos, J.F., Berger, S.L., Dou, Z., Adams, P.D. (2020) Mitochondria-to-nucleus retrograde signaling drives formation of cytoplasmic chromatin and inflammation in senescence. Genes and Development. 34, 428-445. doi/10.1101/gad.331272.119.
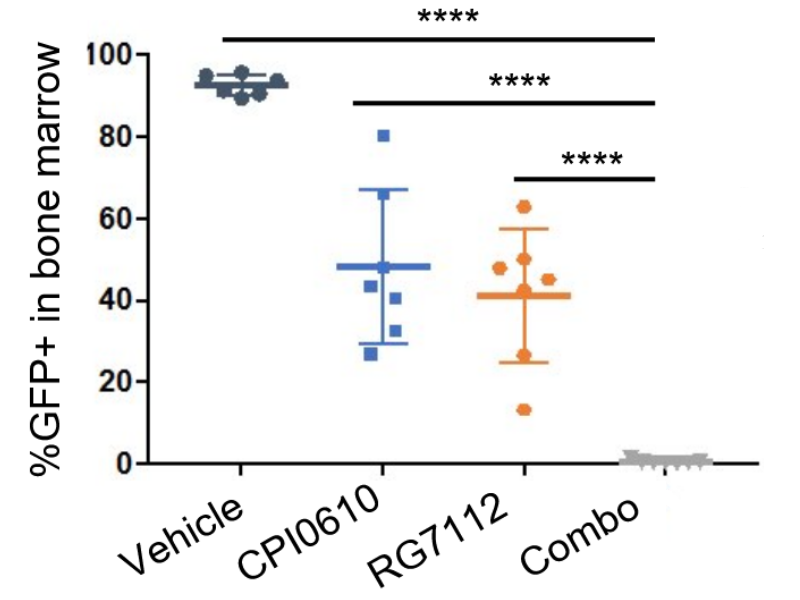
We have performed mechanistic analyses of approved and novel candidate drug therapies for neoplastic disease. The lab has a long-standing interests in melanoma and acute myeloid leukemia (AML). We showed that in oncogene-expressing melanocytes a low level of activated Wnt signaling promotes benign nevus formation (Pawlikowski et al., 2013). However, a high level of activated Wnt signaling, caused by germline sequence variants, promotes giant congenital nevi in the form of congenital melanocytic nevus (CMN) syndrome (Pawlikowski et al., 2015). In a mouse model that closely recapitulates the human genetics, we showed that activated Wnt signaling and an activated Ras oncogene (NRasQ61K) cooperate to drive CMN syndrome, and this is suppressed by acute post-natal treatment with MEK inhibitors (Pawlikowski et al., 2015). Based on these studies our collaborator Dr. Veronica Kinsler is testing MEK inhibitors in babies afflicted by CMN syndrome. In AML, we have investigated the mechanism of action of single epigenetic drug therapies and novel combinations, specifically 5-azacytidine and a BET inhibitor/MDM2 inhibitor combination, CPI0610 and RG7112 (see Figure) (Lund et al., 2014 and Latif et al., 2021).
- Pawlikowski, J.S., Brock, C., Chen, S.C., Al-Olabi, L., Nixon, C., McGregor, F., …, Adams, P.D. (2015). Acute Inhibition of MEK Suppresses Congenital Melanocytic Nevus Syndrome in a Murine Model Driven by Activated NRAS and Wnt Signaling. J Invest Dermatol 135, 2093-2101. PMCID: PMC4539947
- Pawlikowski JS, McBryan T, van Tuyn J, Drotar ME, Hewitt RN, Maier AB, King A, Blyth K, Wu H, Adams PD. Wnt signaling potentiates nevogenesis. Proc Natl Acad Sci U S A. 2013 Oct 1;110(40):16009-14. doi: 10.1073/pnas.1303491110. Epub 2013 Sep 16. PMID: 24043806; PMCID: PMC3791768.
- Lund K, Cole JJ, VanderKraats ND, McBryan T, Pchelintsev NA, Clark W, Copland M, Edwards JR, Adams PD. DNMT inhibitors reverse a specific signature of aberrant promoter DNA methylation and associated gene silencing in AML. Genome Biol. 2014 Aug 30;15(8):406. doi: 10.1186/s13059-014-0406-2. PMID: 25315154; PMCID: PMC4165364.
- Latif AL, Newcombe A, Li S, Gilroy K, Robertson NA, Lei X, Stewart HJS, Cole J, Terradas MT, Rishi L, McGarry L, McKeeve C, Reid C, Clark W, Campos J, Kirschner K, Davis A, Lopez J, Sakamaki JI, Morton JP, Ryan KM, Tait SWG, Abraham SA, Holyoake T, Higgins B, Huang X, Blyth K, Copland M, Chevassut TJT, Keeshan K, Adams PD. BRD4-mediated repression of p53 is a target for combination therapy in AML. Nat Commun. 2021 Jan 11;12(1):241. doi: 10.1038/s41467-020-20378-8. PMID: 33431824; PMCID: PMC7801601.
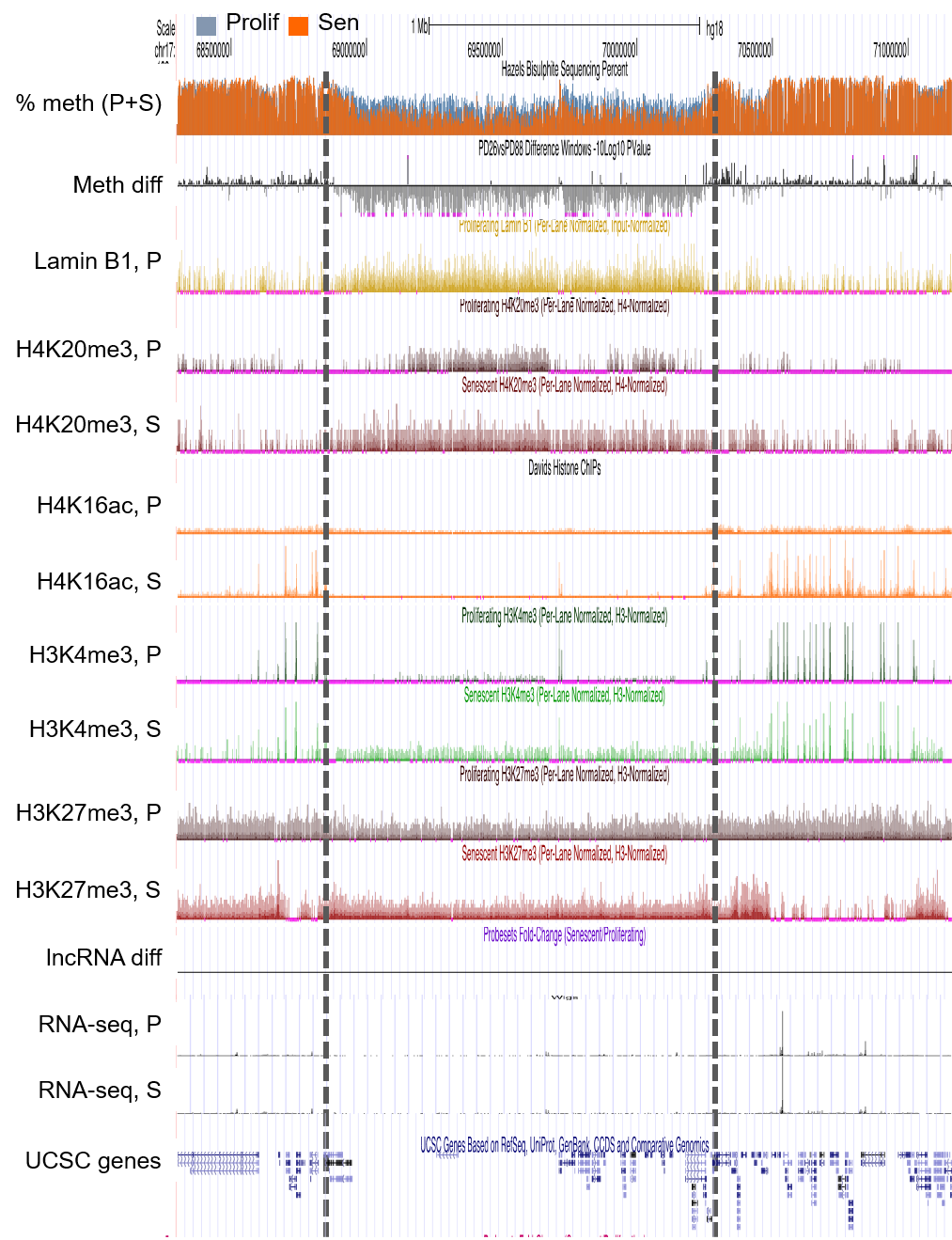
We have mapped the epigenetic landscape of senescent cells, and are defining the causes and consequences of this altered landscape. Cellular senescence is an irreversible proliferation arrest and pro-inflammatory phenotype triggered in primary cells by activated oncogenes and other molecular stresses. As a profound change in cell phenotype, initiation and maintenance of senescence depends on reprogramming of chromatin, the epigenome and gene expression. Cellular senescence is a potent tumor suppressor mechanism. However, accumulation of senescent cells with age also causes tissue aging, by blocking cell and tissue renewal and driving chronic inflammation. Indeed, in contrast to its acute tumor suppressive effects, chronic accumulation of inflammatory senescent cells is tumor promoting. In collaboration with Dr. Shelley Berger, we have mapped the distribution of several critical epigenetic regulators in proliferating and senescent cells, including DNA methylation, several histone modifications, histone variants and nuclear lamins. These collaborative studies have yielded critical insights into gene regulation in senescent cells (Cruickshanks et al., 2013; Rai et al., 2014; Shah et al., 2013), the tumor suppressive and pro-aging effects of senescent cells (Cruickshanks et al., 2013; Nelson et al., 2016).
- Ye X, Zerlanko B, Kennedy A, Banumathy G, Zhang R, Adams PD. Downregulation of Wnt signaling is a trigger for formation of facultative heterochromatin and onset of cell senescence in primary human cells. Mol Cell. 2007 Jul 20;27(2):183-96. doi: 10.1016/j.molcel.2007.05.034. PMID: 17643369; PMCID: PMC2698096.
- Cruickshanks HA, McBryan T, Nelson DM, Vanderkraats ND, Shah PP, van Tuyn J, Singh Rai T, Brock C, Donahue G, Dunican DS, Drotar ME, Meehan RR, Edwards JR, Berger SL, Adams PD. Senescent cells harbour features of the cancer epigenome. Nat Cell Biol. 2013 Dec;15(12):1495-506. doi: 10.1038/ncb2879. Epub 2013 Nov 24. PMID: 24270890; PMCID: PMC4106249.
- Rai TS, Cole JJ, Nelson DM, Dikovskaya D, Faller WJ, Vizioli MG, Hewitt RN, Anannya O, McBryan T, Manoharan I, van Tuyn J, Morrice N, Pchelintsev NA, Ivanov A, Brock C, Drotar ME, Nixon C, Clark W, Sansom OJ, Anderson KI, King A, Blyth K, Adams PD. HIRA orchestrates a dynamic chromatin landscape in senescence and is required for suppression of neoplasia. Genes Dev. 2014 Dec 15;28(24):2712-25. doi: 10.1101/gad.247528.114. PMID: 25512559; PMCID: PMC4265675.
- Shah PP, Donahue G, Otte GL, Capell BC, Nelson DM, Cao K, Aggarwala V, Cruickshanks HA, Rai TS, McBryan T, Gregory BD, Adams PD*, Berger SL*. Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev. 2013 Aug 15;27(16):1787-99. doi: 10.1101/gad.223834.113. Epub 2013 Aug 9. PMID: 23934658; PMCID: PMC3759695. * Co-Corresponding Authors
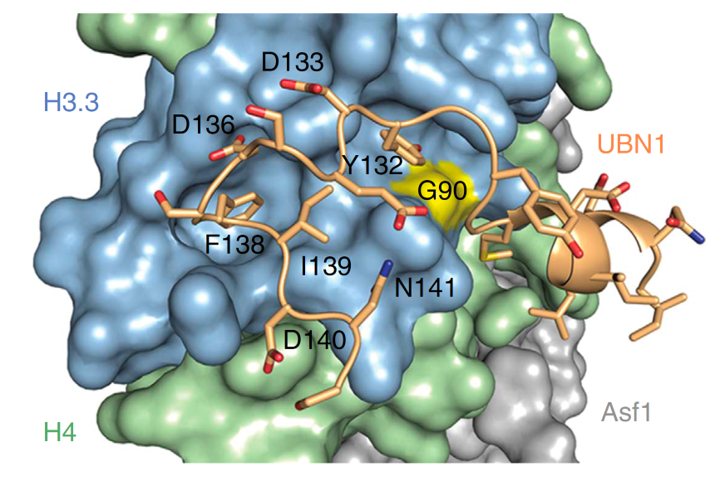
Landmark structure and functional studies on the HIRA histone chaperone complex and its role in senescence-mediated tumor suppression. In collaboration with Dr. Ronen Marmorstein in Philadelphia we have dissected the structure-function relationships between HIRA and its binding partners, UBN1, CABIN1 and ASF1a and substrate histone H3.3 (Zhang et al., 2005). This included a crystal structure of the HIRA/ASF1a interaction surface and more recently the UBN1/histone H3.3 interaction surface (Tang et al., 2006). We were the first to describe the distribution of the HIRA complex across the mammalian epigenome (Pchelintsev et al., 2013). In functional studies, we have demonstrated the role of this DNA replication independent histone chaperone complex in control of chromatin in non-proliferating senescent cells (Rai et al., 2014). These studies have been facilitated by the mouse monoclonal and rabbit polyclonal antibodies that we have made to all subunits of the complex. More recently, we have generated the first conditional knock out mice of HIRA, UBN1 and CABIN1 and are using these to establish in vivo functions (Rai et al., 2014). Of particular note, we have revealed a function for HIRA in promoting healthy aging and suppression of cancer (Rai et al., 2014).
- Zhang R, Poustovoitov MV, Ye X, Santos HA, Chen W, Daganzo SM, Erzberger JP, Serebriiskii IG, Canutescu AA, Dunbrack RL, Pehrson JR, Berger JM, Kaufman PD, Adams PD. Formation of MacroH2A-containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Dev Cell. 2005 Jan;8(1):19-30. doi: 10.1016/j.devcel.2004.10.019. PMID: 15621527.
- Tang Y, Poustovoitov MV, Zhao K, Garfinkel M, Canutescu A, Dunbrack R, *Adams PD, *Marmorstein R. Structure of a human ASF1a-HIRA complex and insights into specificity of histone chaperone complex assembly. Nat Struct Mol Biol. 2006 Oct;13(10):921-9. doi: 10.1038/nsmb1147. Epub 2006 Sep 17. PMID: 16980972; PMCID: PMC2933817. *Co-corresponding authors.
- Pchelintsev NA, McBryan T, Rai TS, van Tuyn J, Ray-Gallet D, Almouzni G, Adams PD. Placing the HIRA histone chaperone complex in the chromatin landscape. Cell Rep. 2013 Apr 25;3(4):1012-9. doi: 10.1016/j.celrep.2013.03.026. Epub 2013 Apr 18. PMID: 23602572; PMCID: PMC3974909.
- Rai TS, Cole JJ, Nelson DM, Dikovskaya D, Faller WJ, Vizioli MG, Hewitt RN, Anannya O, McBryan T, Manoharan I, van Tuyn J, Morrice N, Pchelintsev NA, Ivanov A, Brock C, Drotar ME, Nixon C, Clark W, Sansom OJ, Anderson KI, King A, Blyth K, Adams PD. HIRA orchestrates a dynamic chromatin landscape in senescence and is required for suppression of neoplasia. Genes Dev. 2014 Dec 15;28(24):2712-25. doi: 10.1101/gad.247528.114. PMID: 25512559; PMCID: PMC4265675.
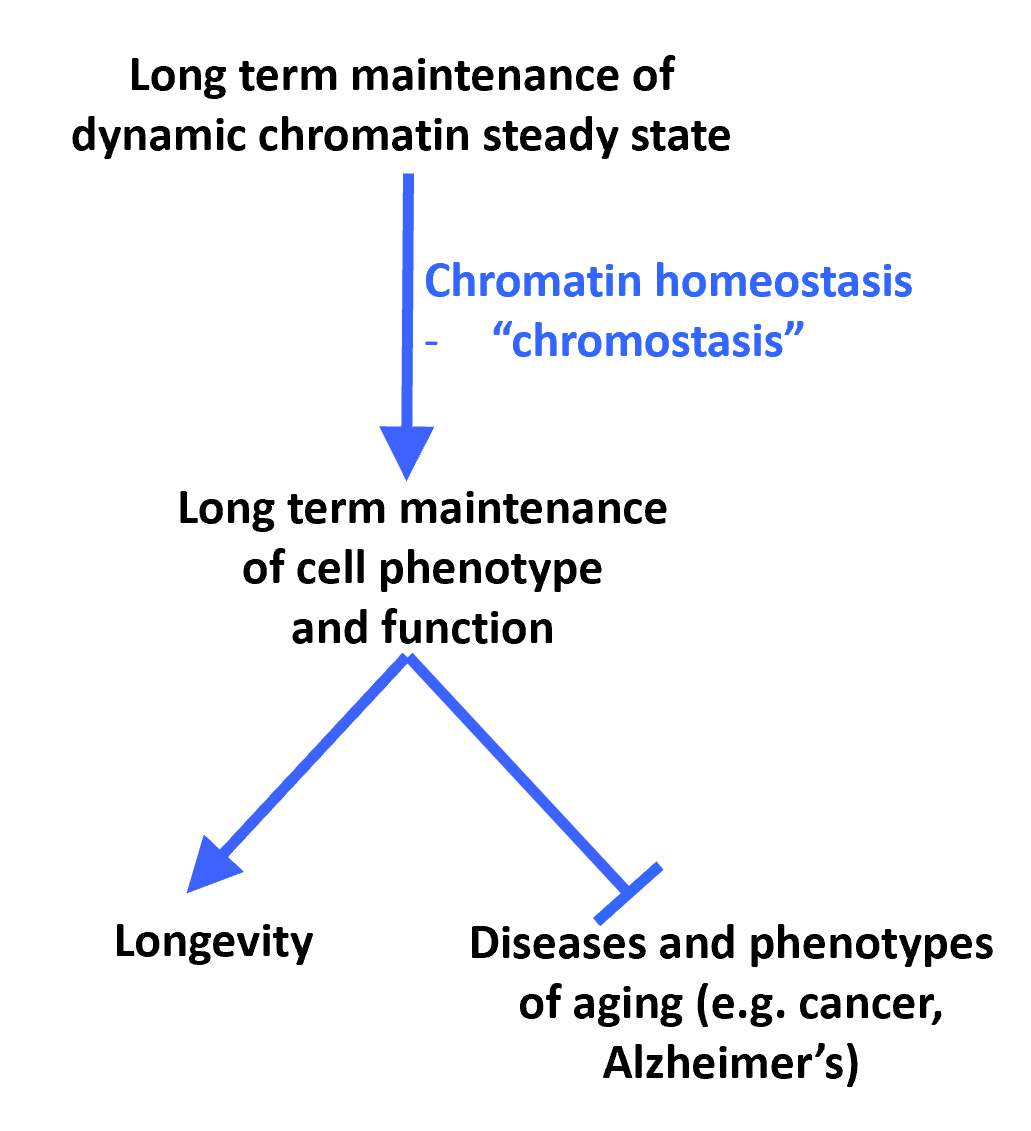
We coined the term "chromostasis" to describe the presumptive homeostatic mechanisms that confer epigenetic stability over the lifecourse, and we were major contributors to the first demonstration of a DNA methylation clock in the mouse. Maintenance of cell phenotype and suppression of disease, including cancer, over the lifecourse depends on a high level of epigenetic stability. However, since chromatin is inherently dynamic (Rai et al., 2014), this steady state stability likely reflects a challenge for the cell. Therefore, presumptive "chromatin homeostasis" or "chromostasis" mechanisms are predicted to actively maintain an epigenetic steady state over the lifecourse, thereby suppressing age-associated disease (Rai et al., 2014). We have shown that histone chaperone HIRA is one such factor that contributes to epigenetic stability in non-proliferating cells (Rai et al., 2014). Recently, we reported the 1st DNA methylation clock in the mouse, and showed that diverse interventions - genetic, dietary and drug - that promote longevity of mice also suppress age-associated epigenetic changes and slow progression of this DNA methylation "clock", i.e. enhance chromostasis (Cole et al., 2017; Wang et al., 2017; Field et al., 2018).
- Rai TS, Cole JJ, Nelson DM, Dikovskaya D, Faller WJ, Vizioli MG, Hewitt RN, Anannya O, McBryan T, Manoharan I, van Tuyn J, Morrice N, Pchelintsev NA, Ivanov A, Brock C, Drotar ME, Nixon C, Clark W, Sansom OJ, Anderson KI, King A, Blyth K, Adams PD. HIRA orchestrates a dynamic chromatin landscape in senescence and is required for suppression of neoplasia. Genes Dev. 2014 Dec 15;28(24):2712-25. doi: 10.1101/gad.247528.114. PMID: 25512559; PMCID: PMC4265675.
- Cole JJ, Robertson NA, Rather MI, Thomson JP, McBryan T, Sproul D, Wang T, Brock C, Clark W, Ideker T, Meehan RR, Miller RA, Brown-Borg HM, Adams PD. Diverse interventions that extend mouse lifespan suppress shared age-associated epigenetic changes at critical gene regulatory regions. Genome Biol. 2017 Mar 28;18(1):58. doi: 10.1186/s13059-017-1185-3. PMID: 28351383; PMCID: PMC5370462.
- Wang T, Tsui B, Kreisberg JF, Robertson NA, Gross AM, Yu MK, Carter H, Brown-Borg HM, Adams PD, Ideker T. Epigenetic aging signatures in mice livers are slowed by dwarfism, calorie restriction and rapamycin treatment. Genome Biol. 2017 Mar 28;18(1):57. doi: 10.1186/s13059-017-1186-2. PMID: 28351423; PMCID: PMC5371228.
- Field AE, Robertson NA, Wang T, Havas A, Ideker T, Adams PD. DNA Methylation Clocks in Aging: Categories, Causes, and Consequences. Mol Cell. 2018 Sep 20;71(6):882-895. doi: 10.1016/j.molcel.2018.08.008. PMID: 30241605; PMCID: PMC6520108.
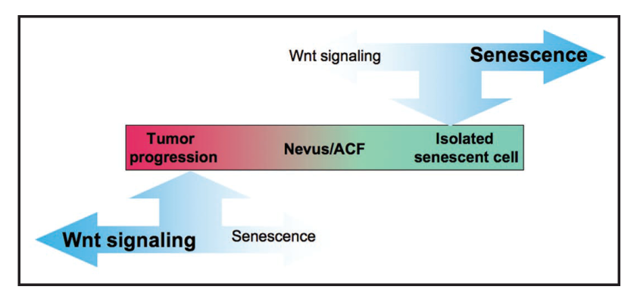
We demonstrated a "tug of war" between tumor suppressive oncogene-induced senescence and oncogenic activated Wnt signaling in melanocytic neoplasia (Adams and Enders, 2008; Ye et al., 2007). The balance between these tumor suppressive and oncogenic activities determines the efficiency of senescence-mediated tumor suppression. For example, we showed that in oncogene-expressing melanocytes a low level of activated Wnt signaling promotes benign nevus formation (Pawlikowski et al., 2013). However, a high level of activated Wnt signaling, caused by germline sequence variants, promotes giant congenital nevi in the form of congenital melanocytic nevus (CMN) syndrome (Pawlikowski et al., 2015). In a mouse model that closely recapitulates the human genetics, we showed that activated Wnt signaling and an activated Ras oncogene (NRasQ61K) cooperate to drive CMN syndrome, and this is suppressed by acute post-natal treatment with MEK inhibitors (Pawlikowski et al., 2015). Based on these studies our collaborator Dr. Veronica Kinsler is preparing to test MEK inhibitors in babies afflicted by CMN syndrome.
Ye X, Zerlanko B, Kennedy A, Banumathy G, Zhang R, Adams PD. Downregulation of Wnt signaling is a trigger for formation of facultative heterochromatin and onset of cell senescence in primary human cells. Mol Cell. 2007 Jul 20;27(2):183-96. doi: 10.1016/j.molcel.2007.05.034. PMID: 17643369; PMCID: PMC2698096.
Cruickshanks HA, McBryan T, Nelson DM, Vanderkraats ND, Shah PP, van Tuyn J, Singh Rai T, Brock C, Donahue G, Dunican DS, Drotar ME, Meehan RR, Edwards JR, Berger SL, Adams PD. Senescent cells harbour features of the cancer epigenome. Nat Cell Biol. 2013 Dec;15(12):1495-506. doi: 10.1038/ncb2879. Epub 2013 Nov 24. PMID: 24270890; PMCID: PMC4106249.
Rai TS, Cole JJ, Nelson DM, Dikovskaya D, Faller WJ, Vizioli MG, Hewitt RN, Anannya O, McBryan T, Manoharan I, van Tuyn J, Morrice N, Pchelintsev NA, Ivanov A, Brock C, Drotar ME, Nixon C, Clark W, Sansom OJ, Anderson KI, King A, Blyth K, Adams PD. HIRA orchestrates a dynamic chromatin landscape in senescence and is required for suppression of neoplasia. Genes Dev. 2014 Dec 15;28(24):2712-25. doi: 10.1101/gad.247528.114. PMID: 25512559; PMCID: PMC4265675.
Shah PP, Donahue G, Otte GL, Capell BC, Nelson DM, Cao K, Aggarwala V, Cruickshanks HA, Rai TS, McBryan T, Gregory BD, Adams PD, Berger SL. Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev. 2013 Aug 15;27(16):1787-99. doi: 10.1101/gad.223834.113. Epub 2013 Aug 9. PMID: 23934658; PMCID: PMC3759695.